Coronavírus
Anvisa diz que ainda faltam dados do Butantan para pedido de uso emergencial de vacina; análise da Fiocruz segue para a próxima fase
Agência informou que já fez duas reuniões com o Butantan neste sábado (9) para tratar da questão. Instituto ficou de complementar dados ‘com brevidade’









Coronavírus
OMS declara fim da pandemia da Covid-19
Os especialistas da Organização Mundial da Saúde chegaram à conclusão de que o vírus não representa mais uma ameaça sanitária internacional.

Nesta sexta-feira (5), a OMS (Organização Mundial da Saúde) anunciou o fim da pandemia da Covid-19. De acordo com seus especialistas, o vírus não representa mais uma ameaça sanitária internacional. Portanto, a emergência da pandemia está oficialmente declarada como encerrada. Ainda assim, segundo o diretor-geral da OMS, Tedros Ghebreyesus, o anúncio não significa que o vírus desapareceu. Atualmente, ainda há…
Coronavírus
Governo libera vacina bivalente contra a Covid-19 para todos acima de 18 anos
Vacinação com a bivalente é ampliada após atingir apenas 16% do público-alvo nas primeiras etapas da campanha.
Na noite desta segunda-feira (24), o Ministério da Saúde anunciou a liberação da vacina bivalente de reforço contra Covid-19 para qualquer pessoa acima de 18 anos de idade. Podem receber a bivalente quem já recebeu, pelo menos, duas doses de vacinas monovalentes, como Coronavac, Astrazeneca ou Pfizer. A aplicação da bivalente deve acontecer em um intervalo de pelo menos quatro…
Coronavírus
Governo de SP derruba obrigatoriedade do uso de máscara no transporte em todo o Estado
A medida vale para trens, ônibus intermunicipais e metrôs de todo o Estado de São Paulo.

Nesta semana, o Governo de São Paulo retirou a obrigatoriedade do uso de máscaras no transporte público em todo o Estado. Assim, a partir de agora, não é mais obrigatório utilizar o item de segurança em ônibus intermunicipais, trens e metrô. Ainda assim, para ônibus municipais, a decisão partirá de cada prefeitura. A medida acontece um dia depois da Anvisa…
Coronavírus
Covid: Jundiaí abre agendamento para vacina bivalente para idosos e imunossuprimidos
O imunizante é uma versão atualizada dos já existentes contra a Covid-19 e pode oferecer uma proteção ainda maior contra a cepa original e subvariantes.
A Saúde de Jundiaí abrirá novo agendamento para doses de vacina Pfizer bivalente contra a Covid-19, nesta terça-feira (28), às 15h. Idosos com 70 anos ou mais e imunossuprimidos com 12 anos ou mais podem agendar pelo site ou APP Jundiaí. Para tomar a dose bivalente, a pessoa deve ter recebido, no mínimo, duas doses da vacina monovalente (Janssen, Pfizer, CoronaVac ou Astrazeneca).…
Coronavírus
Covid: Saúde amplia vacinação com Pfizer Baby para crianças de 3 a 4 anos
A versão pediátrica do imunizante começa a ser aplicada nessa faixa etária a partir de segunda-feira (13)
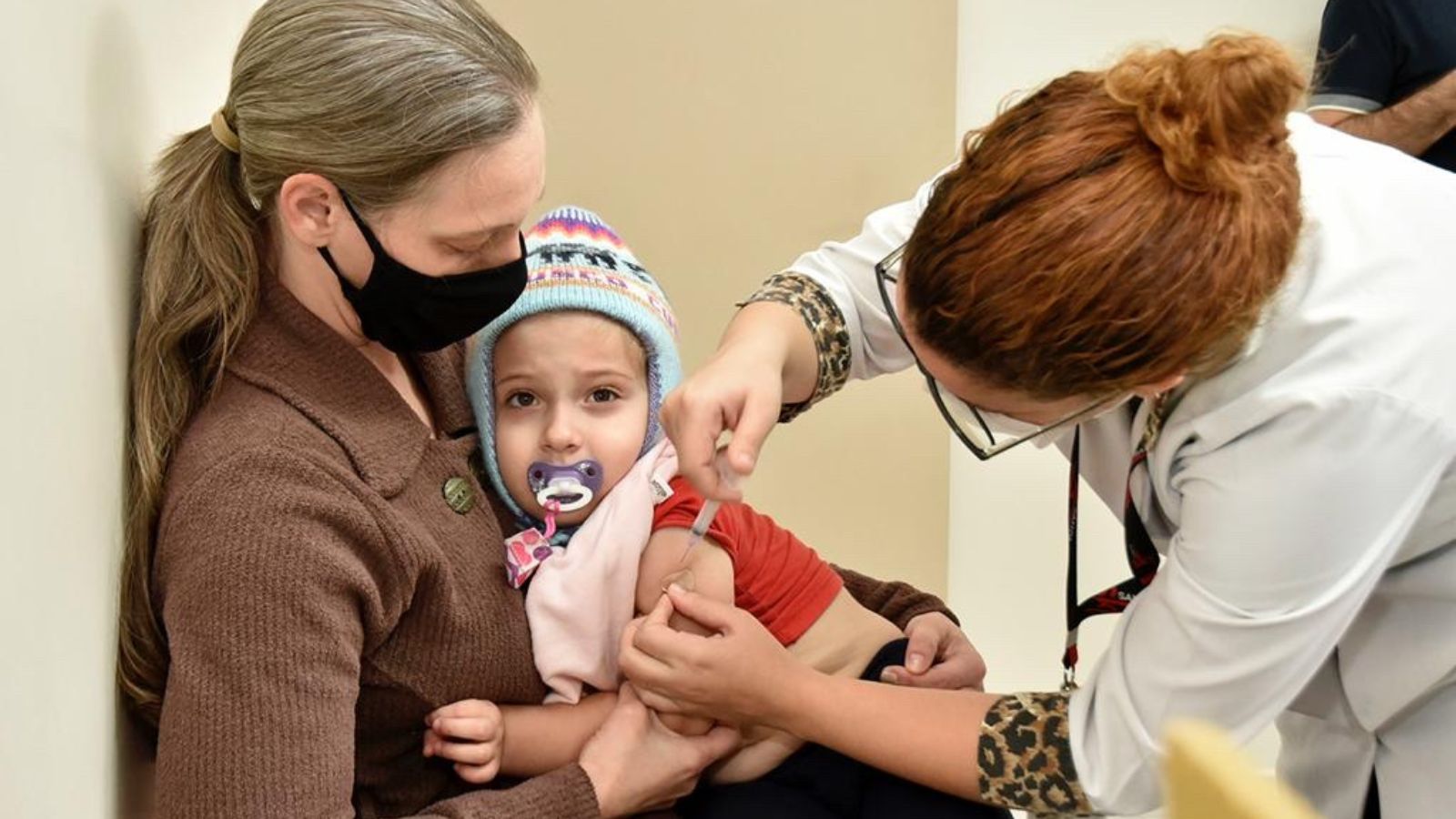
A partir desta segunda-feira (13), Jundiaí passa a aplicar a vacina Pfizer Baby também em crianças de 3 a 4 anos. A princípio, essa versão do imunizante estava disponível apenas para o público de seis meses a 2 anos. Durante a semana, a aplicação ocorre de acordo com calendário de imunização da cidade, disponível no site da Prefeitura de Jundiaí. Em…

Polícia Federal vai investigar postagens com fake news sobre enchentes no Rio Grande do Sul

Saiba como ajudar: entidades de Jundiaí recebem doações para famílias no Rio Grande do Sul

Várzea Paulista colabora com a situação do Rio Grande do Sul; veja como ajudar

É falso que governo gaúcho está fiscalizando documentação de jet skis e barcos que atuam em resgates

Bispo da Diocese de Jundiaí recebe equipe da Assistência Social para alinhamento de trabalhos

Mata-Mato: aprenda a fazer veneno caseiro e elimine as ervas daninhas

3 receitas caseiras de mata-mato para acabar de vez com os invasores

Cozinhar com banha de porco é mais saudável do que com óleos vegetais, diz especialista

Empresa desenvolve canudos comestíveis para substituir plástico

Carrefour de Jundiaí recebe notificação da Prefeitura: agora só pode vender alimentos

ACE Jundiaí promove Rodada de Negócios em parceria com a Renault Valec

Evento de filiação do PL Várzea Paulista

Valec Renault Jundiaí apresenta o novo Kardian

Colégio Divino Salvador completa 70 anos de história
